常用生物信息学格式介绍
本文最后更新于:2024年6月17日 下午
fasta
fasta (维基百科)格式是最基本的表示序列信息(核苷酸或者蛋白质)的格式。这里简单介绍下,fasta 格式的文件通常后缀名为 .fasta 或者 .fa ,其实这都无所谓,因为都是文本文件。 fasta 格式文件(可以包含多条序列)中的一条序列的通常表示方法如下:
1 | |
其中主要分为两个部分:
- 第一部分是序列的定义行(单行),该行的开头是>符号,紧跟着后面的就是该条序列的名称(具有唯一性,即不能和其它序列同名称),即>号和后面的名称的第一字符间是没有任何空白的。一般第一个空格后面的内容即为可选的描述信息。如上面, gi|129295|sp|P01013|OVAX_CHICK 为序列名称, 而 GENE X PROTEIN (OVALBUMIN-RELATED)则为描述信息。注意:有点软件是把一整行当做名称的,所以在出现错误的时候可以查看下格式是否正确。
- 第二部分就是序列,所有的序列碱基或者氨基酸可以都放在一行存储,也可以多行存储,但是建议大家多行存储且单行长度不超过 80 个字符,因为这样容易阅读。且序列的多行之间不能有空行,序列信息描述的第一行与序列数据的第一行之间不能有空行。其中序列数据主要是按照密码表来表示的,*表示是蛋白质翻译的结束。
多行序列举例如下:
1 | |
fastq
fastq 同样是以文本形式来存储序列信息的格式,后缀名通常为 .fastq 或者 .fq ,但是与 fasta 不相同的是,它除了存储序列本身外还存储了序列中每个单元所对应的质量分数,所以 fastq 格式通常用于高通量测试数据的存储。早期是有 Sanger 机构开发的,但是现在已经演变成一个高通量测序的标准了。
fastq 格式文件中一个完整的单元分为四行,每行的含义如下:
第一行: 以@开头,内容同 fasta 的描述行类似
第二行:具体的碱基序列
第三行:以+开头,后面的内容可以和第一行类似,也什么都没有只留+
第四行:以 ASCII 字符集(分数)编码来表示对应碱基的测序质量
比如下面的这个例子:
1 | |
下面以 Illumina 和 NCBI SRA 两个测序数据来源来讲讲它们之间的区别:
通常我们获取测序数据有两种途径,一种是自己通过仪器测定,一种是在公共数据库中(比如之前说到的 NCBI 中的 SRA 数据库)获取,这两种方式主要是在序列名称的命名上和测序质量表示方式上有所不同。
Illumina 序列名称:
1 | |
上述以:隔开的每个字段的含义如下:
| HWUSI-EAS100R | the unique instrument name |
|---|---|
| 6 | flowcell lane |
| 3 | tile number within the flowcell lane |
| 941 | ‘x’-coordinate of the cluster within the tile |
| 1973 | ‘y’-coordinate of the cluster within the tile |
| #0 | index number for a multiplexed sample (0 for no indexing) |
| /1 | the member of a pair, /1 or /2 (paired-end or mate-pair reads only) |
NCBI SRA 数据库
将测序数据提交到 NCBI 的 SRA 数据库时, SRA 数据库会为每一个样本提供一个编号,一般是 SRRxxxxx ,所以从 SRA 数据库上下载公共的测试数据(原始格式为 .sra , 需特定工具转换为 fastq ),其 fastq 格式文件中每个单元的名称是以 SRA 编号接数字加以区分的。比如下面的这个示例:
1 | |
需要注意的是:当把测序数据上传到 SRA 数据库时,它通常会将表示质量的分数 转换为标准的 Sanger 格式 。
质量分数表示法:
由于测序仪器的不同等因素所以对碱基测序质量的表示方式也不相同,在 Fastq 格式文件中,用 ASCII 码表来表示每个碱基的测序质量,下面介绍几种不同的方案:  其中有五种表示方法,Sanger 的码表范围为 ! 至 I ,其对应的数值为 33-73,如果减去 33(即 Phred + 33 表示法)这个基数则范围转换为 0-40,即如果某一个碱基的测序质量为 ! 则对应的测序质量分数为 0,表示测序质量低。其它几种表示法类似(X,I,J,L)。这里介绍测序质量的表示方法是因为后面有的软件是要指定测序数据的质量表示方法。
其中有五种表示方法,Sanger 的码表范围为 ! 至 I ,其对应的数值为 33-73,如果减去 33(即 Phred + 33 表示法)这个基数则范围转换为 0-40,即如果某一个碱基的测序质量为 ! 则对应的测序质量分数为 0,表示测序质量低。其它几种表示法类似(X,I,J,L)。这里介绍测序质量的表示方法是因为后面有的软件是要指定测序数据的质量表示方法。
gff2
GFF(General Feature Format)是一种用于描述基因或者其它序列元素的文件格式,GFF 有几个版本,早期的第 Version 2 和现在的 Version 3. Version 2 是由 Sanger 机构所制定的,而 Version 3 是由 Sequence Ontology Project 制定。正是由于有统一的格式来表示基因等元素,使得 GFF 格式的文件被广泛的使用与 mapping 与基因组数据可视化方面。 GFF2 文件格式是由 tab 隔开的九列值,每一行的九个字段的含义如下:
1 | |
第一列: reference sequence, 该列表示的是特征元素所在的染色体(或者 scaffold,或者 contig),也就是在基因组中的坐标系统,后续一切的注释信息都是基于此列。
第二列:source,该列表示改行注释信息的来源,比如上述的一行表示该行的 CDS 注释信息来自名为“curated”的注释。
第三列:feature,或者说是 method,type, 表示的是该注释的类型,比如上述表示改行注释为 CDS 信息,可以将 source 和 feature 结合起来描述的更加详细。
第四列:start position,在 reference sequence 上的开始位置(坐标),通常是从 1 为起点而不是 0。
第五列:end position, 在 reference sequence 上的结束位置(坐标),一般是大于 start position 的。
第六列:score, 表示该行 feature 的分数,比如序列相似性等,如果没有对应的分数可以用.代替。
第七列:strand,feature 所在链,+表示正链,-表示负链,.表示不确定或者与链无关。
第八列:phase,与蛋白质编码相关,一般是用于 CDS,值的范围为 0-2,表示编码时阅读框的移动相位。
下面这段描述很详细:
‘0’ indicates that the specified region is in frame, i.e. that its first base corresponds to the first base of a codon. ‘1’ indicates that there is one extra base, i.e. that the second base of the region corresponds to the first base of a codon, and ‘2’ means that the third base of the region is the first base of a codon. If the strand is ‘-‘, then the first base of the region is value of
第九列:group,或者称为 attributes,是用于对改行注释更多的描述,以键值对的形式,比如上面的例子表示该 CDS 是属于名为 R119.7 的 transcript。该列中可以存在多个属性,属性之间是用;隔开的。
对于 GFF 格式的理解主要是集中在最后一列,有以下集中情况:
- 对于单个 feature
1
Chr3 giemsa heterochromatin 4500000 6000000 . . . Band 3q12.1 - 对于属于同一集合的多个 feature
IV curated exon 5506900 5506996 . + . Transcript B0273.1 IV curated exon 5506026 5506382 . + . Transcript B0273.1 IV curated exon 5506558 5506660 . + . Transcript B0273.1 IV curated exon 5506738 5506852 . + . Transcript B0273.1比如上面这个例子就表示这四个 exon 都是属于同一个名为 B0273.1 的 transcript,这是表示一个完整 transcript 结构的最基本要求。
GFF2 还可用于序列比对结果表示等其它方面,这里不做介绍了。
gtf(gff2.5)
GTF(Gene Transfer Format)格式是借鉴于 GFF2 格式,也被称为 GFF2.5,大部分字段的定义是和 GFF2 相同的,只是每行的第九列必须带有如下四个域,具体为 gene_id value; transcript_id value; 这样的设计是为了适应一个基因的多个转录本这种情况。比如下面的这个例子:
1 | |
gff3
GFF2(维基百科)格式早期用的比较多,但是现在用的多的是 GFF3 格式,这也是好多软件所支持的,比如 Gbrowse, Jbrowse 等基因组数据可视化工具。先看下面这个简单的例子:
1 | |
第一行的##gff-version 3 通常是需要的,而且必须是在文件的第一行。
前八列和 GFF2、GFF2.5 类似,但是有几点是要特别注意的,主要是将 GFF3 注释数据用于基因组浏览器时,字段中的一些特殊字符比如空格,> %等都需要使用 URL 编码进行转换才能准确的在 web 中进行展示。
第九列同样是表示 attributes,采用的同样是键值对的形式(tag=value),只是这里有几个特定的键,具体如下:
ID,feature 在整个 GFF3 文件中唯一的标识符;
Name,feature 的名字,不同于 ID,Name 不要求唯一,只是方便用户浏览;
Alias, 相当于 feature 的别名;
Parent,表明该 feature 所属的上一级 feature 的 ID,这种关系可用于 exons-transcripts,transcripts-genes,可以看出一个 feature 可以拥有多个子 feature;
Target, 主要是用于序列比对结果的展示,value 的格式为 target_id start end [strand], 其中如果 target_id 中含有空格则需转换为%20;
后面还有些其它属性比如 Note 等,这里不再做详细描述。
下面再来看下典型的例子:
蛋白质编码基因结构 ``` ctg123 example gene 1050 9000 . + . ID=EDEN;Name=EDEN;Note=protein kinase ctg123 example mRNA 1050 9000 . + . ID=EDEN.1;Parent=EDEN;Name=EDEN.1;Index=1 ctg123 example five_prime_UTR 1050 1200 . + . Parent=EDEN.1 ctg123 example CDS 1201 1500 . + 0 Parent=EDEN.1 ctg123 example CDS 3000 3902 . + 0 Parent=EDEN.1 ctg123 example CDS 5000 5500 . + 0 Parent=EDEN.1 ctg123 example CDS 7000 7608 . + 0 Parent=EDEN.1 ctg123 example three_prime_UTR 7609 9000 . + . Parent=EDEN.1
ctg123 example mRNA 1050 9000 . + . ID=EDEN.2;Parent=EDEN;Name=EDEN.2;Index=1 ctg123 example five_prime_UTR 1050 1200 . + . Parent=EDEN.2 ctg123 example CDS 1201 1500 . + 0 Parent=EDEN.2 ctg123 example CDS 5000 5500 . + 0 Parent=EDEN.2 ctg123 example CDS 7000 7608 . + 0 Parent=EDEN.2 ctg123 example three_prime_UTR 7609 9000 . + . Parent=EDEN.2 ctg123 example mRNA 1300 9000 . + . ID=EDEN.3;Parent=EDEN;Name=EDEN.3;Index=1 ctg123 example five_prime_UTR 1300 1500 . + . Parent=EDEN.3 ctg123 example five_prime_UTR 3000 3300 . + . Parent=EDEN.3 ctg123 example CDS 3301 3902 . + 0 Parent=EDEN.3 ctg123 example CDS 5000 5500 . + 1 Parent=EDEN.3 ctg123 example CDS 7000 7600 . + 1 Parent=EDEN.3 ctg123 example three_prime_UTR 7601 9000 . + . Parent=EDEN.31
2
3
4
5
一个名为 EDEN 的基因拥有三个转录本,分别名为 EDEN.1 EDEN.2 EDEN.3, 每个转录本又有 UTR 和 CDS 等信息。
- 序列比对ctg123 est EST_match 1050 1500 . + . ID=Match1;Name=agt830.5;Target=agt830.5 1 451
ctg123 est EST_match 3000 3202 . + . ID=Match1;Name=agt830.5;Target=agt830.5 452 654ctg123 est EST_match 5410 5500 . - . ID=Match2;Name=agt830.3;Target=agt830.3 505 595
ctg123 est EST_match 7000 7503 . - . ID=Match2;Name=agt830.3;Target=agt830.3 1 504ctg123 est EST_match 1050 1500 . + . ID=Match3;Name=agt221.5;Target=agt221.5 1 451
ctg123 est EST_match 5000 5500 . + . ID=Match3;Name=agt221.5;Target=agt221.5 452 952
ctg123 est EST_match 7000 7300 . + . ID=Match3;Name=agt221.5;Target=agt221.5 953 12531
2
- 定量数据ctg123 affy microarray_oligo 1 100 281 . . Name=Expt1
ctg123 affy microarray_oligo 101 200 183 . . Name=Expt1
ctg123 affy microarray_oligo 201 300 213 . . Name=Expt1
ctg123 affy microarray_oligo 301 400 191 . . Name=Expt1
ctg123 affy microarray_oligo 401 500 288 . . Name=Expt1
ctg123 affy microarray_oligo 501 600 184 . . Name=Expt11
2
3
4
5
6
7- 含 Fasta 格式的 GFF3 格式文件 `##gff-version 3 ctg123 . exon 1300 1500 . + . ID=exon00001 ctg123 . exon 1050 1500 . + . ID=exon00002 ctg123 . exon 3000 3902 . + . ID=exon00003 ctg123 . exon 5000 5500 . + . ID=exon00004 ctg123 . exon 7000 9000 . + . ID=exon00005 ##FASTA >ctg123 cttctgggcgtacccgattctcggagaacttgccgcaccattccgccttg tgttcattgctgcctgcatgttcattgtctacctcggctacgtgtggcta tctttcctcggtgccctcgtgcacggagtcgagaaaccaaagaacaaaaa aagaaattaaaatatttattttgctgtggtttttgatgtgtgttttttat aatgatttttgatgtgaccaattgtacttttcctttaaatgaaatgtaat cttaaatgtatttccgacgaattcgaggcctgaaaagtgtgacgccattc ...` 该 GFF3 文件中含有对应的序列,以##FASTA 作为标示。
## bed
bed([genome 介绍](http://www.genome.ucsc.edu/FAQ/FAQformat.html#format1)、[bedtools 介绍](http://bedtools.readthedocs.org/en/latest/content/general-usage.html)、 [asia 介绍](http://asia.ensembl.org/info/website/upload/bed.html?redirect=no))格式同样是用于展示序列注释信息,有相应的软件来处理这类格式的文件,如 bedtools。可以用在类似 GBrowse 这样的基因组数据可视化工具中。 以 tab 隔开,它必须的三个字段为 chrom、chromStart、chromEnd,还有 9 个可选字段。注意:用于在 GBrowse 上展示相关注释的 bed 格式通常第一行有一个关于 track 的描述信息。
比如下面的例子:
track name=pairedReads description=”Clone Paired Reads” useScore=1
chr22 1000 5000 cloneA 960 + 1000 5000 0 2 567,488, 0,3512
chr22 2000 6000 cloneB 900 - 2000 6000 0 2 433,399, 0,3601
1 | |
:497:R:-272+13M17D24M 113 1 497 37 37M 15 100338662 0 CGGGTCTGACCTGAGGAGAACTGTGCTCCGCCTTCAG 0;==-==9;>>>>>=>>>>>>>>>>>=>>>>>>>>>> XT:A:U NM:i:0 SM:i:37 AM:i:0 X0:i:1 X1:i:0 XM:i:0 XO:i:0 XG:i:0 MD:Z:37
:20389:F:275+18M2D19M 99 1 17644 0 37M = 17919 314 TATGACTGCTAATAATACCTACACATGTTAGAACCAT >>>>>>>>>>>>>>>>>>>><<>>><<>>4::>>:<9 RG:Z:UM0098:1 XT:A:R NM:i:0 SM:i:0 AM:i:0 X0:i:4 X1:i:0 XM:i:0 XO:i:0 XG:i:0 MD:Z:37
:20389:F:275+18M2D19M 147 1 17919 0 18M2D19M = 17644 -314 GTAGTACCAACTGTAAGTCCTTATCTTCATACTTTGT ;44999;499<8<8<<<8<<><<<<><7<;<<<>><< XT:A:R NM:i:2 SM:i:0 AM:i:0 X0:i:4 X1:i:0 XM:i:0 XO:i:1 XG:i:2 MD:Z:18^CA19
:21597+10M2I25M:R:-209 83 1 21678 0 8M2I27M = 21469 -244 CACCACATCACATATACCAAGCCTGGCTGTGTCTTCT <;9<<5><<<<><<<>><<><>><9>><>>>9>>><> XT:A:R NM:i:2 SM:i:0 AM:i:0 X0:i:5 X1:i:0 XM:i:0 XO:i:1 XG:i:2 MD:Z:35
解释:
其中可以看出 Aligenment 2 和 Alignment 3 是成对的 reads,其插入长度为 314。 bam 格式中的 b 是 binary 的意思,是 sam 格式的二进制表示方式,为什么要用二进制表示呢? 因为 sam 格式文件大小通常是十分大的,一般是以 G 为单位,所以为了减少存储量等因素而将 sam 转换为二进制格式以便于分析。
sam/bam 格式是由特定的一些软件(比如 samtools)来处理的,包括格式互转、排序、建立索引、搜寻突变等操作,后续分析中会详细讲解 samtools 工具的使用方法。 
## vcf
[vcf](http://samtools.github.io/hts-specs/VCFv4.2.pdf)(Variant Call Format)格式是用于表示突变信息的文本格式,可以用来表示 single nucleotide variants, insertions/deletions, copy number variants and structural variants 等。VCF 格式同样是分为两大部分,一部分是注释描述信息,一部分是具体的突变信息,其中注释信息是以##开头的,我们来看下面这个例子: 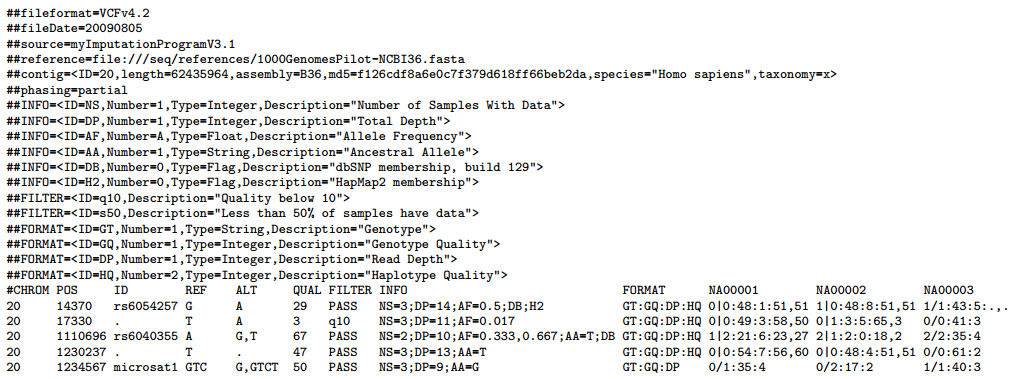 我们着重来关注第二部分的每列字段是什么含义:
CHROM 即 chromosome, 染色体名称;
POS 即 position, 发生突变的参考序列的位置(从 1 开始计数);
ID 突变的名称;
REF 参考序列 POS 上的碱基;
ALT 发生突变的碱基,多个的话以,连接, 可选符号为 ATCGN\*,大小写敏感;
QUAL 基于 Phred 格式的表示 ALT 的质量,也可以理解为可靠性;
FILTER 过滤后的状态,即按照可靠性进行筛选;
INFO 额外信息,可结合注释描述信息进行理解  针对 vcf 格式有如 bcftools 等软件进行处理。
---

微信支付

支付宝